Other two principal and unique STING Millennium features are:
|
The main feature of the STING Millennium is the ability to combine data delivery through the web with structural analysis tools in order to provide a self-contained instrument for macromolecular studies. |
|
STING Millennium is both
didactic tool as well as research tool.
It is easy to use and requires virtually no training time. |
| STING Millennium
allows one to load a PDB file (molecular structure) and to receive
information about underlying molecular sequences. This is one of three KEY
features of the STING
Millennium: Other two principal and unique STING Millennium features are: |
STING Millennium is composed by two main windows. The Sequence Window displays sequence and contains the general menus with the commands. The Structure (or Graphics) Window displays the macromolecular rendered tree-dimensional structure.
In general terms STING Millennium provides the following services:
|
|
Ability to easily select residues in the sequence, select elements of secondary structure, as well as offer a wide variety of methods for rendering and coloring a molecule (mostly available through ACTION menu). |
|
|
Defining 3D neighbors to arbitrary selected residue |
|
|
Definition and display of amino acids participating in interfacial regions between polypeptide chains (through WINDOWS/Interface chain menu selection) |
|
|
Building surfaces of whole molecule or just IFR part of it |
|
|
Interactive Ramachandran plot, permitting rapid identification of residues in the disallowed regions and display of selected residues in the structure window |
|
|
Calculation of residue frequency within selected chain or on interface, as well as frequency of those residues filtered through chosen contact parameters. |
|
|
Hydrogen bond net calculation with special attention given to participation of water molecules. |
|
|
Contacts definition and calculation for the whole molecule and/or interfaces |
|
|
Convenient 2D graphical presentation of parameters extracted from 3D structure |
|
|
Display of sequence neighbors and calculation of relative sequence conservation for the family of homologous proteins. |
In the links entry in the main menu, several external services
that deal with PDB files are listed. These consist of links to web sites containing
programs that accept a PDB code as input to perform useful tasks, which makes
STING Millennium highly integrated with other important data resources.
Activating
all STING Millennium menu options
STING
Millennium Control Panel BASIC Commands
STINGpaint
PDB_Mining
|
|
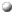 Color Coded Residues: Hydrophobicity/Charge
Instant Display of Residue/Nucleotide
Number Within Sequence Linear Sequence to 3D Fold
STING Millennium Link Secondary structure elements
identified Gaps in Sequence Clearly Indicated
Chains Separated and Displayed
STINGpaint: WWW tool for sequence
and MSA coloring Database Linking
PDB_Mining
Color Coded Residues: Hydrophobicity/Charge
Instant Display of Residue/Nucleotide
Number Within Sequence Linear Sequence to 3D Fold
STING Millennium Link Secondary structure elements
identified Gaps in Sequence Clearly Indicated
Chains Separated and Displayed
STINGpaint: WWW tool for sequence
and MSA coloring Database Linking
PDB_Mining
Sequence color
coding
Sequence Window brings linear protein sequence color coded
with respect to Hydrophobicity and charge groups!
|
Nucleotide sequence is also color coded:
|

The STING Millennium 3D Graphics Window and Sequence Window are interconnected ;
|
What is the correct action sequence necessary to obtain desired rendering in STING Millennium 3D window?:
OBSERVE resulting rendering in STING Millennium 3D window! |
|
Important note:
|
The user can place the mouse over the single letter code in the sequence
and ask question such as: where in the 3D fold is this residue placed?
|
JMOL users - when STING operates in Jmol mode, the REFRESH button has a delay of up to 3-4 seconds before causing any action in Jmol image. This is internal characteristics of Jmol and is only noticable on first click at the Refresh button. If the user desires to restart STING Millennium 3D display with one of the FOUR available whole protein display styles (wireframe [1], cartoon [2], backbone [3] and ribbon [4]), REFRESH button should be used. Pressing consecutively REFRESH button, will show available display choices from which the user might start NEW molecule rendering, using above described action sequence. |
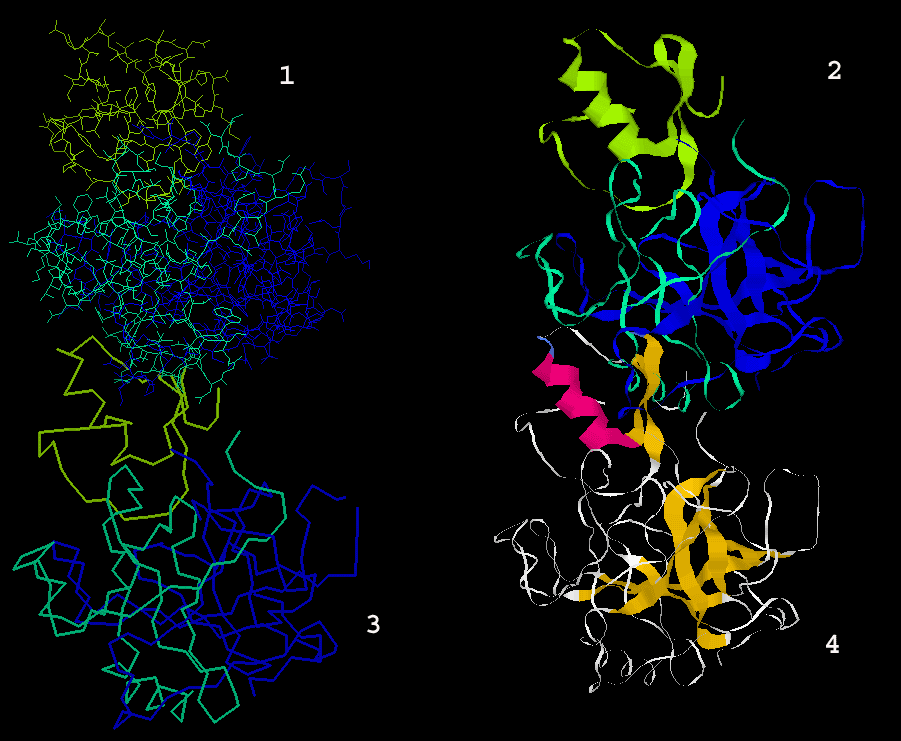
Secondary structure
regions
Similarly, the user can slide the mouse over the secondary structure colored
bars Helices (red lines below
the sequence) and Extended Sheets (blue
lines below sequence) and see on STING Millennium Status Frame the
sequence region covered by this element of the secondary structure.
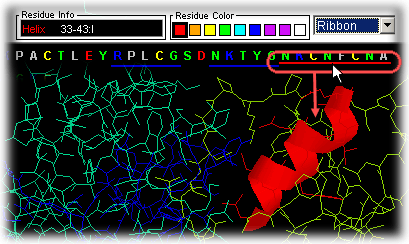
Sequence Gaps in PDB
file
Gaps are clearly indicated in the Sequence window by use of
"-----" in place of residue/nucleotide single letter code, for indication
of residue missing at that position. This is also one of the key features
of STING Millennium presentation.

Sequence Chains
in PDB file
Chains are also clearly indicated in the Sequence Window so
that the user can have access to residues separated by distinct chain identifier!
This key feature is very handy, once a user would like to examine an
interface between two protein chains; digital access to residues (belonging
to different chains) can then produce graphical CPK/WS/.. positions of critical
residues in the protein fold.
Warning: residue number is separated by ":" from the Cain identifier.
Chain identifier can be either letter or number, or first letter of any 3-letter
code used in PDB file as chain identifier! For more details, user should consult
PDB file formats!

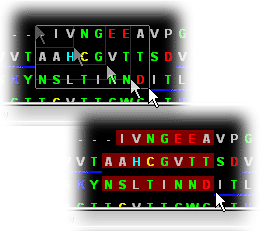
Here we will only describe basic commands from the STING
Millennium control menu. There are 8 menu options on STING
Millennium sequence window:
|
Picking
|
Surface
|
Links
|
Help
|
| Clear All
|
HOH off
|
| Wireframe
|
Ligand on
|
| Ribbon |
Ligand off
|
| Color CPK
|
Ligand Pocket
|
| Color by Chain
|
HOH + Ligand
|
| Color by Structure
|
|
| HOH on |
STING Millennium Control Menu and Commands
Note: Some of the commands presented here are simple copy of the
standard CHIME commands. We have chosen to put them in the Action menu
options for the easier access. Most usable STING
Millennium commands are actual scripts, made to facilitate molecular
structure and macromolecular interface analysis. We found "Interface
on", "Ligand Pocket", "HOH+Interface", "HOH+ligand",
Interface: 1st/2nd half and "Charged Residues" very conveniently,
one stroke apart from viewing on the Graphics Window.
Clear All
This is the only way to refresh the graphics frame (if starting from
scratch is desired).
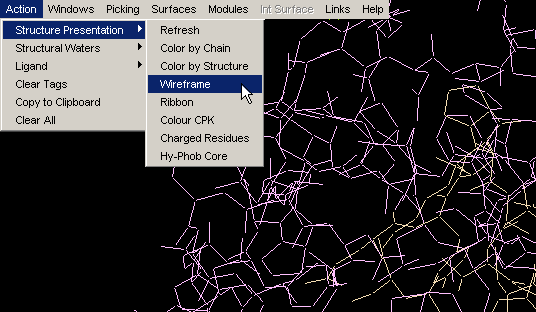
Wireframe
If your graphics screen becomes too cluttered, use Clear All and then
restart by using the Wireframe command. You will end up with (you guessed
right!) wireframe representation of the molecule.
Note: if previously you have used "Color by chain", these color codes
are retained, which we found useful once you enter into the chain of analysis
procedures. If you really desire to start with original CPK colors, use
option "Color CPK" in addition to "Clear All" + "Wireframe"! {Example used
here is 1ppf.pdb}
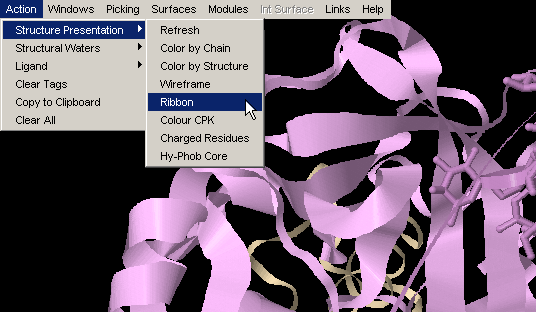
Ribbon
Ribbon simply turns on ribbon around protein main chain backbone. {Example
used here is 1ppf.pdb}
Color CPK
This option is made available for the single purpose of convenience:
easy, one stroke action to obtain, for example, Interface built in earlier
analysis, color coded CPK (instead of color coded with respect to chain
identifier, e.g.)! {Example used here is 1ppf.pdb}
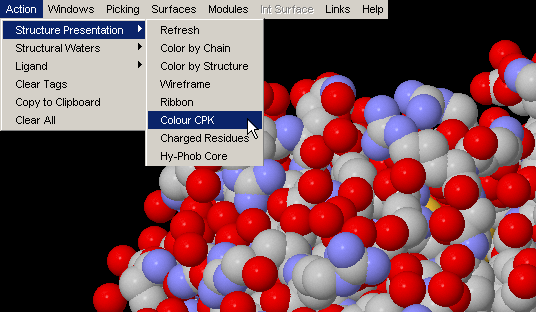
Color by Chain
Convenient coloring by Chain also gives you opportunity to see if any
chain is actually broken by introduction of the gap in the sequence (this
info, however, could be easily observed by looking at the Sequence Window.)
{Example used here is 1ppf.pdb}
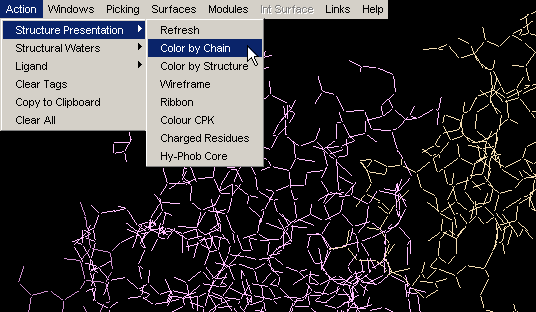
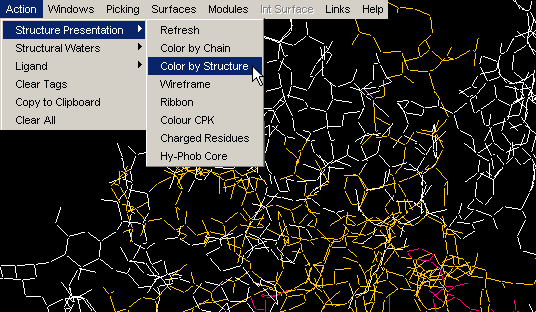
Color by Structure
This command colors the molecule by protein secondary structure.
Alpha helices are colored magenta, [240,0,128], beta
sheets are colored yellow, [255,255,0], turns
are colored pale blue, [96,128,255] and all other residues are colored white.
The secondary structure is either read from the PDB file (HELIX and SHEET
records), if available, or determined using Kabsch and Sander's DSSP algorithm.
Note:regions of protein fold colored by this command DO NOT
NECESSARILY coincide with Secondary Structure identifiers (red and blue
lines within the Sequence Window). Difference originates in
calculated versus indicated nature of two approaches, respectively. This
is very nicely compared by Protein
Dossier presentation.{Example used here is 1ppf.pdb}
HOH on
STING Millennium default Graphics
Window, comes with crystal waters turned on. If present, these water
molecules might make further analysis difficult, as they might clog the
picture. In this case, use command "HOH off". Later in an analysis, one
might wish to turn them on, for examination of their presence within interface
layer, for example. Once you use "HOH on" button, water molecules will
change from default red color to magenta!
This is done in order to facilitate visual identification of HOH molecules,
especially in cases when default HOH color, red, is also used by other chain
or ligand!
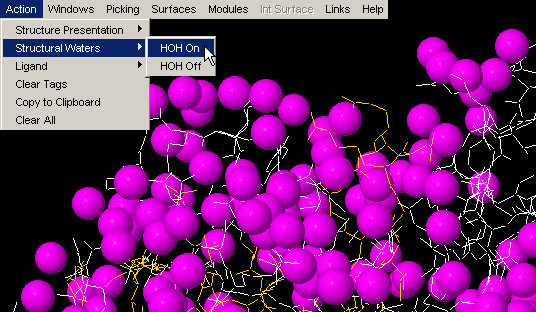
HOH off
STING Millennium default Graphics
Window, comes with crystal waters turned on. If present in large
number, waters should be removed from the visual using this command!
Ligand on
This command will turn visual presentation of any present ligands
on.
If you do not see single atom HETATOMs such as Cu, Mg, Mn, Zn, Fe, Cl, Ca,
Br etc., see here how to
proceed!
Ligand off
Somewhat surprisingly, this command, really turns ligand visual off.
:)
Ligand Pocket
"Ligand Pocket" will erase anything else from the graphics frame but
ligand and residues side_chain atoms in contact with it! Contact is defined
by distance, set to 4.0 Angstrom, and measured between ligand atoms and
any other atoms belonging to ligand surrounding chains! This feature is
very convenient, once you turn on the wireframe display of the rest of the
molecule, by using "Wireframe" command. Position of the Ligand and
surrounding molecules will be very clearly displayed for further analysis.
Combine this with Ligand on/off, Color by chain and get better
insight into ligand 3D environment! Also, very much used in combination
with HOH + Ligand!
Note: distance of 4.0 Angstroms will pretty much satisfy requirements
for both hydrogen bond formation, as well as for hydrophobic interactions!
Note #2: there is a TUTORIAL with
worked example for this usuful STING option!
If you see number of residues being indicated as a POCKET, but no LIGAND
to which they are associated, this does not necessarily means that STING
made a mistake. It only reflects limitation of the script we used to paint
multi-atom LIGANDS and not single atom ones. See here
how to proceed.
HOH + Ligand
This command will have visualized Ligand and water molecules in contact
with it. It is often necessary to have this information on contacting water
molecules around the ligand for proper H-bond counting. Some structural
water molecules are identified easily in this way!
Contact between ligand and HOH molecules is defined here by distance, set
to 3.3 Angstroms, and measured between ligand atoms and any other HOH molecule
(actually, in most cases, an Oxygen atom).
Note: distance of 3.3 Angstroms is considered maximum distance between
Hydrogen donor and acceptor, that could still bring about hydrogen bond
formation.
Charged Residues
This command will visualize all charged residues using van der Waals
dotted surface. All lysines and arginines
will be blue color coded, aspartic acid and
glutamic acid will be red and histidine
will be color coded cyan. This is a very useful feature, once you would
like to know charge distribution in vicinity of ligand or maybe at molecular
interface!
Note: this command will visualize (turn on) all charged residues,
irrespective to the chain identifier. We found this convenient in most cases,
as user can get CPK presentation of charged residues in the chain of interest,
and dot color-code of charge residues in contact with interface in focus
(and not belonging to the same chain) - therefore having glimpse of complementarity)!
{Example used here is 1ppf.pdb}
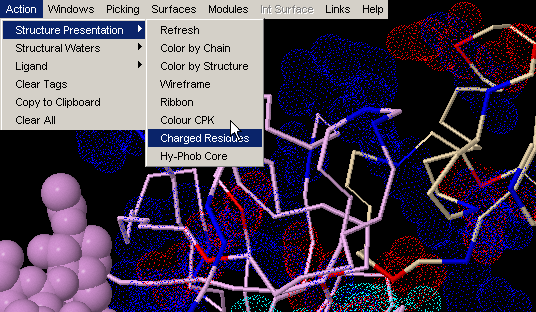
This command will visualize all hydrophobic residues (supposedly located in the core of the protein) in CPK presentation and also all charged residues using van der Waals dotted surface. {Example used here is 1ppf.pdb}

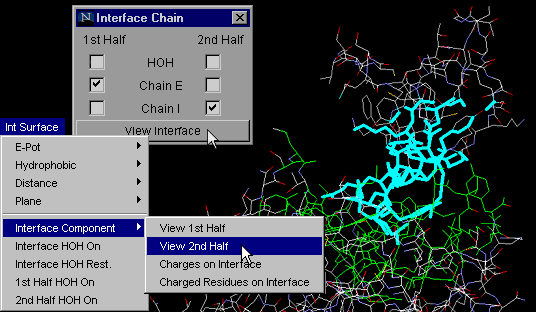
Windows & Int Surface Menu Options:
Interface on
Interface HOH on
Interface Component : View 1st
Half
Interface Component: View 2nd Half
1st half HOH
2nd half HOH
Interface on
Obviously, this command only functions for the pdb files with more than
one chain. For the list of all files with more than one chain, the user
can consult our PDB_Mining.
To generate image bellow, we used 1ppf.pdb. In green
thick wireframe is the interface part (1st half) that belongs
to the E chain and in thick cyan wireframe
is the interface part that belongs to the I chain. {Example used here is
1ppf.pdb}
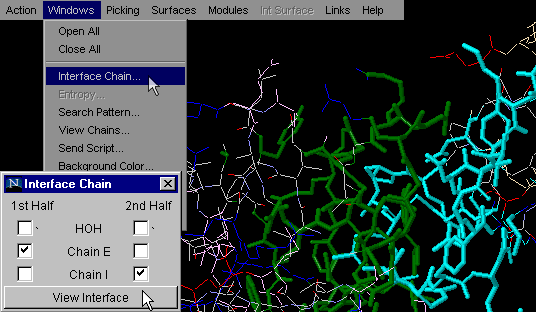
This is one of the most used commands in STING
Millennium . What we like about it is the simplicity
of getting general information on interfaces between two molecular
chains, all in one mouse stroke. This command will turn on only atoms at
an INTERFACE of the first two chains in a PDB file. The choice of chains
for which user desires to see Interface, is the essential for proper interface
building for all pdb files with more than 2 chains.. In combination with
command "Color by chain" (issued prior to Interface on) graphical
information is even more emphasized.
Note:Interface is defined based on a distance, set to 8.0 Angstroms,
and measured between any two atoms in different chains! A value of 8.0 Angstroms
was chosen empirically; we first tried a distance of two times 3.3 Angstroms
(Hydrogen acceptor from one chain, to water molecule, to hydrogen donor
on the other chain). This would be distance of 6.6 Angstroms, but we found
that graphical presentation of the interfaces is much more "complete" if
distance of 8.0 Angstroms is used. We judged completeness by how well an
interface is populated by atoms, or how many holes we have on the chains
interface.
Obviously, the user should consider Interface graphics presentation more
as a guide, than as a exact interface definition.
| As a matter of fact, the exact Interface
Forming Residue (IFR) ensemble is defined by
our algorithm in FORMIGA: there,
we define IFR as those residues that have different solvent
accessibility in isolation and in complex. The option to use is: "Show
Interface Area". Also, the exact Interface definition, based
on Buried surface area upon complex formation,is available in our
package: HORNET.
|
Interface HOH on
This command is very useful for analysis of the interfaces and
water molecules captured between Interface Forming Residues (IFR).
Availability of such quick identification of these waters may aid in
identification of indirect H-bond formation between two chains (with
involvement of structural water molecules).
Note:HOH molecules visualized by this command are identified
here by a distance, set to 3.3 Angstroms, and measured between a subset
of atoms belonging to the interface from one chain, to any HOH molecule
(actually, in most cases, an Oxygen atom); This is then done for the
other chain (its IFR and HOH molecules at defined distance of 3.3 Angstroms).
Finally presented waters are actually intersection of water molecule
ensembles defined above! In other words, the only water molecules presented
(color coded magenta), are those that satisfy the geometric condition
of being 3.3 Angstrom (maximum) distance from both chains! These HOH
molecules are likely to make H-bonds with both chains, contributing
effectively to the energy of binding!
Note: distance of 3.3 Angstroms is considered here as the maximum
distance between a Hydrogen donor and acceptor that still defines a
hydrogen bond.
These water molecules are then easily observable if you use option:
"Interface Component: View 1st Half" and "Interface Component: View
2nd Half". One can actually analyze only one chain IFR, with
HOH molecules which are likely to make H-bonds with both chains!
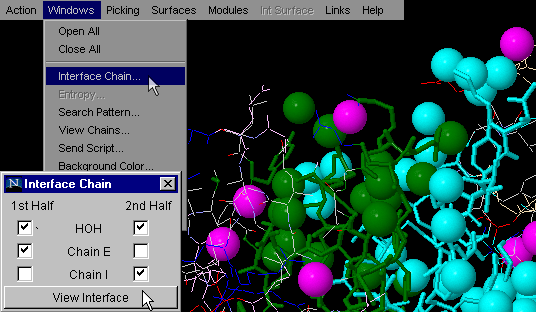
{Example used here is 1ppf.pdb}
Interface Component:
View 1st Half
This particular command will allow the user to observe only one
of the two chains forming a macromolecular interface. This option allows
the user to examine in detail only half of a complementary surface (see
example in tutorial section!).
As explained above for "Interface on", using the "Interface Component:
View 1st Half" button one can actually analyze only one chain IFR,
with HOH molecules which are likely to make H-bonds with both chains!
{Example used here is 1cho.pdb}
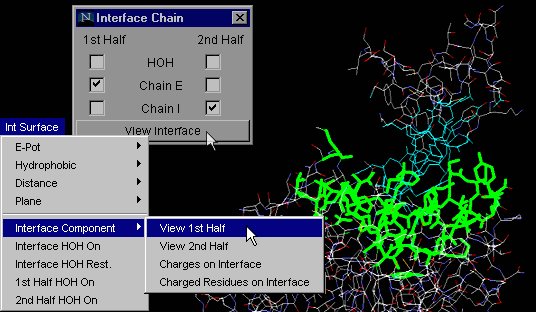
Interface Component:
View 2nd Half
Same as command "Interface Component: View 1st Half", but obviously
tuned for visualization of the second half of the complementary surfaces.
See tutorial for the real power of these two last commands!
Note:As explained above for "Interface on", with "Interface Component:
View 2nd Half" button one can actually analyze only one chain IFR,
with HOH molecules which are likely to make H-bonds with both chains!
{Example used here is 1cho.pdb}
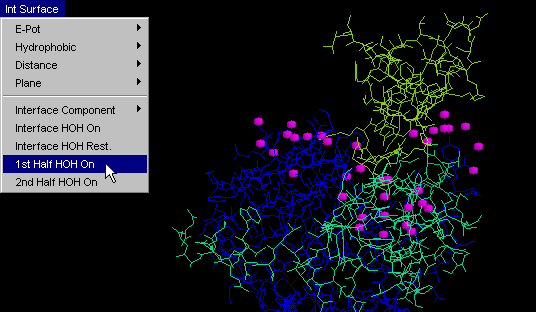
1st half HOH on
This command will conveniently display only one (first) part of
the facing surfaces at molecular interface, in addition to water molecules
which are 3.3 Angstroms distant from any of IFR of that chain.
Difference between this command and "Interface: 1st half" /"Interface:
2nd half" is that the former one will generally show many more HOH molecules
than the latter ones. This is due to the more restrictive condition
imposed for the latter commands, with respect to which water molecules
will be shown. Namely, "HOH+ 1st half" (and so the "HOH+ 2nd half")
will show one chain IFR and all HOH at 3.3 Angstroms from it!
On the other hand, "Interface: 1st half" and "Interface: 2nd half" will
only show those HOH molecules which are 3.3 Angstroms distant from BOTH
chains, which is a much more restrictive condition! User can easily
grasp the difference between two HOH molecule ensembles and conceptualize
the importance of the difference! {Example used here is 1cho.pdb}
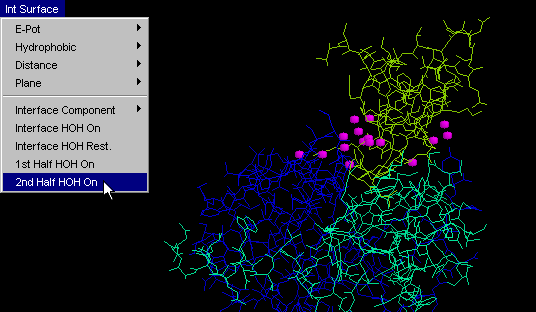
2nd half HOH
Same as "HOH+ 1st half", but obviously tuned for visualization
of another half of the complementary surface. {Example used here is
1cho.pdb}
STINGpaint
STINGpaint was developed to allow the presentation of residue characteristics
in the Sequence Window. As a consequence, during development
of STING project, we have slightly expanded on STINGpaint idea and adopted
it for use with Multiple Sequence Alignment (MSA) coloring. It turns
out that this tool was very interesting for people wanting to easily
grasp specifically colored regions along the MSA. In addition, our STINGpaint
is also a part of our ongoing work for STING-2, a package that will
be able to show both sequence alignments (in the Sequence Window)
and structures (in the Graphics Window) for respective sequences!
STINGpaint now supports following sequence and MSA formats:
1 50
WRP25 SGPWSWCDPA TGYQVSALTG CRAMVKLQCV KSQVPEAVLR DCCQQLADIN
WRP26 SGPWMWCDPA TGYQVSALTG CRAMVKLQCV GSQVPEAVLR DCCQQLADIN
WRP24 SGPWMWCYPG QAFQVPALPA CRPLLRLQCN GCQVPEAVLR DCCQQLAHIS
WRP27 SGPWMWCDPA MGHRVRPLMG CRAMVKLQCV GNQVPEAIQR DCCQELANIT
AI1FAT ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~
AI2FAT ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~
51 100
WRP25 NEWCRCGDLS SMLRSVYQEL GVREGKVLPG CRKEVMKLTA ASVPEVCKVP
WRP26 NEWCRCGDLS SSLRSVYQEL GVREGKVLPG CRKEVMKLTA ASVPEVCKVP
WRP24 NEWCRCG~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~
WRP27 NNWCRCHDLG SMLNSVYQEL GAREGTVFPG CRKEVMKLTV ASVPAVCKVP
AI1FAT ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~
AI2FAT ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~